Analyze Melanoma Patient 78¶
Here we perform analysis on the gene expressions of cells from the melanoma patient number 78. For simplicity we have converted the dataset into TPM. The original count data is available at Gene Expression Omnibus: GSE72056.
Import necessary packages¶
[1]:
%load_ext autoreload
%autoreload 1
[2]:
import sys
import pandas as pd
import numpy as np
import pickle as pkl
import sklearn as skl
import sklearn.preprocessing
import scipy.stats
import matplotlib as mpl
import matplotlib.pyplot as plt
Warning information from TensorFlow may occur. It doesn’t matter.
[3]:
import tensorflow as tf
tf.set_random_seed(1)
import cyclum
from cyclum import writer
/home/shaoheng/.conda/envs/tensorflow-gpu/lib/python3.6/site-packages/h5py/__init__.py:36: FutureWarning: Conversion of the second argument of issubdtype from `float` to `np.floating` is deprecated. In future, it will be treated as `np.float64 == np.dtype(float).type`.
from ._conv import register_converters as _register_converters
[4]:
input_file_mask = 'data/melanoma/M78_tumor'
Read data¶
We do not have cell-cycle labels for the cells any more.
[5]:
def preprocess(input_file_mask):
"""
Read in data and perform log transform (log2(x+1)), centering (mean = 1) and scaling (sd = 1).
"""
sttpm = writer.read_df_from_binary(input_file_mask)
label = pd.read_csv(input_file_mask + '-label.csv', sep="\t", index_col=0)
return sttpm, label
sttpm, label = preprocess(input_file_mask)
There is no convention whether cells should be columns or rows. Here we require cells to be rows.
[6]:
sttpm.head()
[6]:
| C9orf152 | RPS11 | ELMO2 | CREB3L1 | PNMA1 | MMP2 | TMEM216 | TRAF3IP2-AS1 | LRRC37A5P | LOC653712 | ... | GPLD1 | SNORD115-39 | RAB8A | RXFP2 | PCIF1 | PIK3IP1 | SNRPD2 | SLC39A6 | CTSC | AQP7 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cy78-CD45-neg-1-B04-S496-comb | -0.101847 | 0.007892 | 0.904987 | 0.0 | 0.940964 | 0.362364 | 1.468877 | -0.794159 | 0.0 | -0.137973 | ... | -0.443219 | 0.0 | -0.001803 | -0.087706 | -1.099878 | 0.124245 | 0.069167 | 0.333042 | -0.390829 | -0.133081 |
| cy78-CD45-neg-3-H06-S762-comb | -0.101847 | -0.202088 | 0.875485 | 0.0 | 1.228029 | 2.059569 | 1.475918 | -0.433542 | 0.0 | -0.137973 | ... | 0.074614 | 0.0 | 0.658312 | -0.087706 | 0.795460 | 0.865745 | 0.430293 | 0.688241 | -0.973564 | -0.133081 |
| cy78-CD45-neg-1-D07-S523-comb | -0.101847 | 0.048798 | -0.044918 | 0.0 | 0.092909 | -0.693664 | 1.148125 | -0.361802 | 0.0 | -0.137973 | ... | -0.939599 | 0.0 | 0.951421 | -0.087706 | 0.263022 | -0.508476 | 0.559235 | 0.116920 | 0.332409 | -0.133081 |
| cy78-CD45-neg-3-D01-S709-comb | -0.101847 | 0.025618 | -1.046433 | 0.0 | 0.457558 | -0.693664 | 1.070903 | -0.206473 | 0.0 | -0.137973 | ... | -0.432214 | 0.0 | 1.198078 | -0.087706 | 1.066057 | -0.508476 | 0.920172 | 0.153443 | -1.072105 | -0.133081 |
| cy78-CD45-neg-2-B08-S596-comb | -0.101847 | 0.143624 | 1.412362 | 0.0 | 0.253140 | -0.693664 | -0.583134 | -1.203133 | 0.0 | -0.137973 | ... | 1.698251 | 0.0 | 0.974617 | -0.087706 | -0.521885 | 0.187822 | -0.166806 | 0.687519 | -0.361164 | -0.133081 |
5 rows × 23686 columns
[7]:
label.head()
[7]:
| tumor | malignant(1=no,2=yes,0=unresolved) | non-malignant cell type (1=T,2=B,3=Macro.4=Endo.,5=CAF;6=NK) | |
|---|---|---|---|
| cy78-CD45-neg-1-B04-S496-comb | 78 | 2 | 0 |
| cy78-CD45-neg-3-H06-S762-comb | 78 | 2 | 0 |
| cy78-CD45-neg-1-D07-S523-comb | 78 | 2 | 0 |
| cy78-CD45-neg-3-D01-S709-comb | 78 | 2 | 0 |
| cy78-CD45-neg-2-B08-S596-comb | 78 | 2 | 0 |
Set up the model and fit the model¶
Fitting the model may take some time. Using a GTX 960M GPU it takes 6 minutes.
[8]:
model = cyclum.PreloadCyclum(sttpm.values, q_circular=3, q_linear=0)
[9]:
pseudotime, rotation = model.fit()
model.close()
pretrain burnin
step 2000: loss [0.49333668, 27318.037, 75596.17] time 5.09
pretrain train
step 2000: loss [0.49329627, 27318.31, 75596.17] time 3.80
step 4000: loss [0.49329615, 27318.295, 75596.18] time 3.35
midtrain burnin
step 2000: loss [0.49329615, 9224.356, 39934.965] time 9.69
midtrain train
step 2000: loss [0.49329615, 9224.358, 39934.953] time 9.60
step 4000: loss [0.49329615, 9224.36, 39934.95] time 8.79
finaltrain train
step 2000: loss [0.5430872, 9208.43, 22069.293] time 12.77
step 4000: loss [0.5481686, 9208.404, 11894.999] time 11.64
step 6000: loss [0.55096513, 9208.468, 6089.2246] time 11.65
finaltrain refine
step 2000: loss [0.5542275, 9208.411, 4898.9956] time 12.75
step 4000: loss [0.5545521, 9208.403, 3995.3237] time 11.62
step 6000: loss [0.5546467, 9208.404, 3257.5784] time 11.63
step 8000: loss [0.5548571, 9208.413, 2659.4236] time 11.62
step 10000: loss [0.5553257, 9208.426, 2173.033] time 11.63
Full time 135.76
Illustrations¶
We illustrate the results on a circle, to show its circular nature. There is virtually no start and end of the circle. Red, green and blue represents G0/G1, S and G2/M phase respectively. The inner lines represents single cells. The cells spread across the The areas outside
[10]:
%aimport cyclum.illustration
[11]:
color_map = {'tumor': {78: "red"},}
cyclum.illustration.plot_round_distr_color(pseudotime, label['tumor'], color_map['tumor'])
pass

[12]:
sttpm2 = sttpm - np.concatenate([np.cos(pseudotime + i * 2 * np.pi / 3) for i in range(3)], axis=1) @ rotation
Propose genes¶
We show the top 20 proposed gene here.
[13]:
weight = np.sqrt((rotation[0, ] - rotation[1, ] / 2 - rotation[2, ] / 2) ** 2 +
3 * (rotation[1, ] - rotation[2, ]) ** 2 / 4 + 1e-12)
order = list(reversed(np.argsort(weight)))
for i in order[1:20]:
print(sttpm.columns.tolist()[i])
UGDH-AS1
NME1
LOC100131257
FBLIM1
ORC4
ATP5G3
MAB21L3
ABCC9
FABP5
C1QBP
LSM10
REXO1L1
ATP5G1
TOMM5
NDUFA8
TLCD2
S100A11
COX6C
LOC646214
AXL/MITF program¶
We show that cyclum helps clarify AXL/MITF contrast.
[14]:
with open('data/melanoma/AXL_marker_genes.txt') as file:
axl_genes = file.read().splitlines()
with open('data/melanoma/MITF_marker_genes.txt') as file:
mitf_genes = file.read().splitlines()
Corrected data¶
The corrected data follows a better negative correlation.
[15]:
sttpm3 = sttpm2[label['malignant(1=no,2=yes,0=unresolved)'] == 2]
[20]:
axl_score = sttpm3[axl_genes].mean(axis=1) - sttpm3.mean(axis=1)
mitf_score = sttpm3[mitf_genes].mean(axis=1) - sttpm3.mean(axis=1)
plt.scatter(axl_score, mitf_score)
np.corrcoef(axl_score, mitf_score)
[20]:
array([[ 1. , -0.23826302],
[-0.23826302, 1. ]])
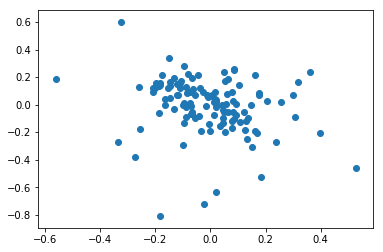
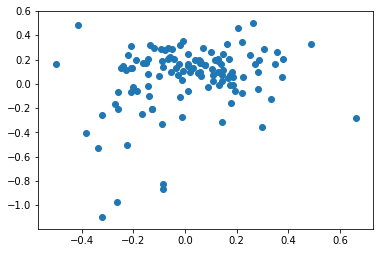
Original data¶
[19]:
sttpm4 = sttpm[label['malignant(1=no,2=yes,0=unresolved)'] == 2]
axl_score = sttpm4[axl_genes].mean(axis=1) - sttpm4.mean(axis=1)
mitf_score = sttpm4[mitf_genes].mean(axis=1) - sttpm4.mean(axis=1)
plt.scatter(axl_score, mitf_score)
np.corrcoef(axl_score, mitf_score)
[19]:
array([[1. , 0.23976131],
[0.23976131, 1. ]])
[ ]: